医疗器械的电子副本提交程序 行业和食药监督管理指南

医疗器械的电子副本提交程序
行业和食品药品监督管理局工作人员指南
文件发布日期:2012年12月31日
该文件草案发布于2012年10月17日
有关本文件的任何疑问,请联系CDRH器械评审办公室 301-7966055,或CBER交流、推广和发展办公室1-800-835-4709 或 301-827-1800。
美国卫生与公众服务部
食品药品监督管理局
器械与放射健康中心
生物制品评估和研究中心
前言
公共评论
书面意见和建议可随时向文档管理部(HFA-305), FDA,5630Fishers Lane, rm. 1061, Rockville, MD 20852提交,供部门审议;电子版意见可提交http://www.regulations.gov。所有意见都应该使用在联邦公报上公布的可用性通知的案卷编号来标识。可能直到文件下次修订或更新时,评论才会被机构受理。
其他副本
其他副本可从互联网获得。贵公司还可以发送电子邮件请求本指南的电子副本:dsmica@fda.hhs.gov,或向301-847-8149发送传真请求以接收复印件。在贵公司提出请求时,请使用本指南的文档编号(1797)以供识别。
本指南文件的其他副本还可获自生物制品评估和研究中心(CBER)的交流培训和企业协助办公室(HFM-40),1401Rockville Pike, Suite 200N, Rockville, MD 20852-1448,或向1-800-835-4709或301-827-1800电话索取,邮件:ocod@fda.hhs.gov,或登录网站:http://www.fda.gov/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/default.htm.
医疗器械的电子副本提交程序
产业和食品药品监督管理局工作人员指南
1. 简介
本指南的目的是解释医疗器械新的电子副本(eCopy)提交程序。食品药品监督管理局安全和创新法案(FDASIA)(出版物L. 112-144)第1136节,增补了联邦食品、药品和化妆品法案(FD&C法案)第745A(b)节,要求提交eCopy并发布该最终指南。本指南介绍了食品药品监督管理局(FDA)如何根据FD&C法案第745A(b)执行eCopy程序,包括eCopy预期将提高审评过程的效率,允许可对电子版本进行立即审评,而不仅仅依赖于纸件版本。
除了其他方面,本指南还特别提供了FD&C法案第745A(b)(2)(A)规定的有效的eCopy标准。根据第745A(b),本最终指南中确定的申请类型必须包含提交符合本指南标准的eCopy的程序和受理,以供FDA审评,除非它们被认定为可豁免的或可免除的。未提交eCopy或提交的eCopy不符合本指南中的标准,则提交的申请会被暂停,直到提交了有效的eCopy并经FDA确认符合标准,已经获得豁免或免除的申请除外。在提交的申请被暂停时,评审计时将不会开始,也不会对提交的申请进行审评。
在第745A(b)节中,国会规定了标准、免除条件和指南中所述的豁免,并授予FDA明确的法定授权,以实施法定的eCopy要求。因此,本文件是根据“FD&C法案”第745A(b)节(即,标准、免除条件和豁免)规定了此项要求,表明,通过使用必须或必需这些词,该文件无需受制于FDA的良好指南实践(GGP)法规中的常规限制,例如指南不能设立法律上强制责任的要求,参见21 CFR 10.115(d)。
然而,本文件还就FDA对eCopy法定要求的解释,以及监管机构目前关于实施eCopy程序其他方面的最佳方法的考虑提供了指导。因此,在本文件的范围中包括了并非在745A(b)(2)项下规定的“标准”,“免除条件”或“豁免”的条款,本文件不会为任何人创造或赋予任何权利,也不会约束FDA或公众,而只是代表了该监管机构目前对于该项主题的考量。在本指南的这些部分中使用了“应”,表示建议或推荐的内容,但不是必需的。如果替代方法可以满足适用的法规和规章的要求,则可以使用该替代方法。如果贵公司想讨论替代方法,请联系负责实施本指南的FDA工作人员。如果无法确定合适的FDA工作人员,请拨打本指南标题页上列出的电话号码。
为遵守GGP法规并确保受监管方和公众了解该指南文件不具有约束力,FDA指南通常会包含标准语言,以解释指南应仅视为建议,除非引用了特定的法规或法定要求。FDA未在本指南中包含这个标准语言,因为它不是对本指南所有作用的准确描述。本指南同时包含了有约束力和无约束力的条款,指南中只要是根据“FD&C法案”第745A(b)节规定的“标准”,“免除条件”和“豁免”,就具有约束力。
eCopy程序并不意图影响(减少或增加)申请人提交的用以支持认证或批准的数据类型或数量[1],申请内容请参阅其他的FDA器械或CDRH程序特定的指导文件。(http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/default.htm)和CBER(http://www.fda.gov/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances / General / ucm214106.htm)。
[1]出于该指南的目的,申请人包括“提交者”、“申办方”、或“持有者”。
2. 什么是eCopy?
电子副本(eCopy)被定义为一种纸质提交资料的精确副本,在压缩盘(CD),数字视频光盘(DVD)或闪存盘上创建和提交。eCopy是带有签名的附函[1]和提交的完整文件的纸质副本[2]。
注意,仅包含有纸质提交资料中某些部分的单独CD(例如,在某些附件中将数据列表加入到套表中的CD)将不再被接受,因为这样的CD不能满足本指南所述的有效eCopy的要求。必须按照上述eCopy的定义提交CD,以便使申请获得受理。
[1]附函签名应使用水笔(即,墨水)签名或有效的数字签名。请注意在IDE或上市前申请(如,510(k)s的真实性和准确性声明)的其他形式提交中,也需使用水笔签名或数字签名。
[2] eCopy不能被认为是一个电子形式的申请, eSubmissions的相关信息,请参见“FDAeSubmitter”(http://www.fda.gov/ForIndustry/FDAeSubmitter/default.htm) 和“Regulatory Submissions inElectronic Format for BiologicProducts”
(http://www.fda.gov/BiologicsBloodVaccines/DevelopmentApprovalProcess/ucm163685.htm).
3. eCopy和纸质提交资料内容之间的差异是否可被接受?
虽然eCopy被定义为纸质副本的精确复制件,但在有限的情况下,eCopy和纸质副本之间的差异可能是合理的,因为纸质副本不能或不适合分析目的(例如,便于生物研究监测检查的原始数据和统计分析程序[1],数据行列表)或不可行(例如,视频,x射线)。eCopy的关键属性是它必须以电子形式包含该申请类型要求的所有数据[2],换句话说,eCopy必须包含FDA评审所需的所有信息,而纸质副本中可以包含一个交叉引用eCopy中某些信息的位置占位符。
附件1中的附函必须包含eCopy的声明,并说明纸质版本和eCopy之间的任何差异。如果eCopy中包含了纸质副本中未纳入的信息,则纸质副本中必须有一个占位符,以指示审评员进行重新定向(例如,有一页陈述来指示评审员找到eCopy中的特定部分)。
FDA考虑将eCopy和附函加载入相应中心的官方文档库中,作为官方记录。eCopy和纸质版本之间任何未公开的差异,会被要求纠正并可能延迟对该申请的审评。
4. 什么申请类型需要提交eCopy?
根据FDASIA第1136节增补的FD&C法案第745A(b)条,需要提交以下申请类型的eCopy:
上市前通告提交(510(k)s),包括第三方510(k)s;
自动III类器械认定申请的评价(新申请);
上市前批准的申请(PMA),包括过渡期PMA;[3]
[1]关于电子提交的信息,请参见“上市前申请的临床数据”(http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/HowtoMarketYourDevice/PremarketSubmissions/ucm136377.htm).
[2]例如,510(k) 申请的内容要求请见21 CFR 870.87和807.92; 首次PMA申请的内容要求请见21 CFR 814.20。
[3]包括所有PMA申请类型,包括,但不限于,首次PMSs,panel-track补充申请,180-天补充申请,生产场地变更补充申请,30-天通知,135-天补充申请,和上市后研究的补充申请,也包括涉及联系人或所有者或所有关系变更和要求增加范围的修订申请。
模块化PMAs;
产品开发方案(PDPs);
临床研究用器械的豁免(IDEs)[见下文的部分豁免];
人道主义的器械豁免(HDEs),包括人道主义用途器械的认定要求(HUD);
紧急使用授权(EUAs)[1] [见下文的豁免];
某些临床研究用新药的申请(INDs);[2]
某些生物制品许可证的申请(BLAs);[3]和
预申请.[4]
在首次申请后,所有后续申请所提交的eCopies,包括修订资料(修订包括增加文件和CLIA分类增加文件)、补充资料和上述申请类型的报告[5]也需要提交,即使是在eCopy的要求实施前向FDA提交了首次申请。
提交文件的大小没有要求,无论是以单页提交(即附函是唯一的内容)还是以多卷提交,eCopy的要求均适用。
对缺陷信的回复需要正式提交给CDRH或CBER的文件控制中心[6](DCC),以作为修正稿或补充资料录入,因此受制于eCopy的要求。
请注意,一旦申请提交并已在审评,eCopy的要求不适用于互动审评过程(通过电子邮件,电话和/或传真)获得信息,如果该信息未提交给CDRH或CBER的DCC。但是,如果申请人选择向CDRH’s或CBER’s DCC提交对互动审评要求的回复(仅会发生在该回复文件的大小无法通过电子邮件或传真沟通时),它将作为修正稿录入并受制于eCopy的要求。
豁免
如上所述,FDA确定了援引至法律的受制于eCopy要求的申请类型。然而,法律还允许FDA制定豁免于eCopy要求的标准。因此,由于以下申请类型可能存在的紧急性质,FDA认为这些可以豁免eCopy的要求:
两种特定类型的IDE – 关怀使用IDE申请和紧急使用IDEh申请[7];和
所有EUAs。
虽然根据该项豁免,这些申请类型不要求eCopies,但FDA鼓励这些申请在可行时提交eCopies,以利于审评的过程。如果贵公司选择提交eCopy,则必须符合附件1[8]中列出的标准。
免除
FDA认为,鉴于软件的广泛应用而得以低成本或无成本地创建可接受的eCopy,所有申请人应该有能力提供eCopy。
[1]更多关于EUAs的信息请参见题为“医疗用品的紧急使用授权(http://www.fda.gov/RegulatoryInformation/Guidances/ucm125127.htm).
[2]仅适用于那些在公共卫生署(PHS)法案第351节项下列为生物制品并受CBER监管的医疗器械,也需要在BLA申请前先提交IND申请。这些器械通常预期用于筛选输血性传染病的供体血液。
[3]仅适用于那些在PHS法案第351节项下列为生物制品并受CBER监管的医疗器械,其不需要在BLA申请前先提交IND申请。这些器械通常包括那些用于检测输血性药物的供体/受体相容性的试剂。
[4]请参见指南草案,题为“医疗器械: 申请前程序和与FDA人员的会议。(http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/ucm310375.htm).
[5]报告,包括所有适用于一种申请类型的所有报告,包括年度/定期报告和上市后报告。FD&C法案第745A(b)节不适用于 21 CFR Part 803项下的医疗器械报告提交。
[6]对于CDRH和CBER的DCC地址,请参见21 CFR 807.90。
[7]更多关于关怀使用IDE和紧急使用IDE申请的细节,请参见CDRH的器械建议页面,“IDE Early/Expanded Access”(http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/HowtoMarketYourDevice/InvestigationalDevi ceExemptionIDE/ucm051345.htm#compassionateuse)和FDA的“IDEs政策和程序指南”(http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/ucm080202.htm)。
[8]如果提交eCopy不可行,但是有相关的电子资料,如影像学数据对纸质副本中的资料进行补充,请联系审评部门以了解如何提交这些资料。
5. 是否有其他不受制于eCopy法规的申请类型可以提交eCopies?
尽管根据FD&C法案第745A(b)节,不要求eCopy,但FDA也接受并强烈建议贵公司提交以下资料的eCopies:
主访问文件(MAF);
513(g)要求的信息(513(g)s);和
临床实验室改进法案(CLIA)分类 - 器械申请豁免(CLIA“X”文件)。
这三种申请类型的eCopies提交是自愿的; 但是,如果贵公司选择提交eCopy,则必须满足附件1中列出的标准。
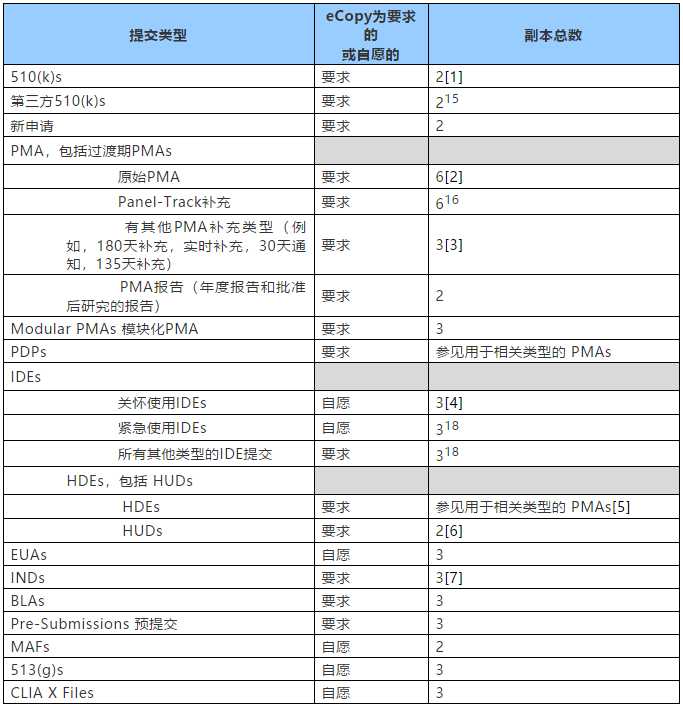
6. 需要提交多少份副本?
eCopy程序不会改变提交给FDA的总份数,下表1中提供了与每种申请类型相关的副本数,其中eCopy是必需的或自愿的。
表1-提交的副本数量
[1]参见 21 CFR 807.90(a)(3)(c).
[2]参见21 CFR 814.20(b)(2).
[3]参见21 CFR 814.39(c).
[4]参见 21 CFR 812.20(a)(3).
[5]参见 21 CFR 814.104(b)(4).
[6]参见 21 CFR 814.102(d).
[7]参见 21 CFR 312.23(d).
对于那些要求提交eCopy的申请类型,eCopy(含签名的附函和适当的eCopy声明)将作为所要求的副本数量中的一份,用于该申请类型。FDA会接受额外的eCopies代替额外的纸质副本,只要和eCopy一起提交至少一份纸质副本即可,所需副本的总数保持不变。
对于仅需要提交两份副本的申请类型,一份必须是eCopy,另一份必须是纸质副本。对于提交需要多于两个副本的申请类型,此政策允许提交的申请形式有更多的灵活性。例如,对于首次PMA,应提交:(1)一份eCopy和五份纸质副本;(2)五份eCopies和一份纸质副本;或(3)任何其他组合,副本总数为六个,其中只要有至少一个eCopy和一个纸质副本。[1]
提交关怀使用IDE申请,紧急使用IDE提交,EUAs,MAFs,513(g)s,和CLIAX文件时,可以选择仅提交纸质副本。但是,如果上述申请选择提交eCopy,请参阅以上详细信息。
7. eCopy的处理步骤是什么?
以下是提交和接受eCopy的处理步骤。
a. eCopy的标准是什么?
关于提交给FDA的eCopy的标准,请参考附件1。不符合附件1中标准的eCopy将无法被加载系统接受。
b. 在将eCopy发送给FDA之前,如何知道eCopy是否符合标准?
当前尚无FDA工具可用以预先验证申请人已经完成的eCopy。但是,在FDA的网站http://www.fda.gov/ForIndustry/FDAeSubmitter/ucm317334.htm上提供了一个新的免费的eSubmitter-eCopies工具,我们强烈建议申请人使用。该工具的使用是可选的;然而,该工具的好处之一是它实时创建了一个符合附件1中描述的标准eCopy。使用eSubmitter-eCopies工具旨在防止因需要解决技术问题而使贵公司提交的申请延迟审评。
注意,该工具提供格式化的eCopy内容,贵公司可以下载到本地驱动器,然后刻录到CD,DVD或闪存盘。不能通过FDA的网关传输eCopy,若在生成eCopy时有任何技术问题,请在向FDA提交eCopy之前联系cdrhesub@cdrh.fda.gov。
[1] FDA 审评员不会向申请人要求额外的eCopy或纸质副本。
c. 如果涉及第三方该怎么办?
在第三方(例如律师事务所,顾问)代表申请人提交申请的情况下,eCopy仍必须满足eCopy的标准,以便成功处理,无论是由贵公司(申请人)还是由提交方完成。虽然申请人的附函可以包括或不包括在eCopy内,但我们的标准要求提交方应包括一份带有eCopy声明的签名附函,如附件1所述。
d. 如果这是第三方510(k)怎么办?
在生成第三方510(k)时涉及两个不同的当事方:(1)认证人和(2)申请人,每一方都受制于eCopy的要求。因此,每一方(即认证人和申请人)必须提供自己的:
符合附件1标准的eCopy(单个CD,DVD或闪存盘),和
如附件1所述的带有eCopy声明的签名附函。
因此,第三方510(k)将提供两个单独的eCopies,鉴于此,每个eCopy必须清楚地标记,以示其属于认证人还是申请人。
FDA同意申请人在向FDA提交第三方510(k)之前多次与认证人进行互动,无论在向FDA提交第三方510(k)前互动的次数如何,申请人的eCopy必须刻录在单个的CD、DVD或闪存盘上,以便于我们的软件加载。例如,申请人可以将每轮互动整理为eCopy的不同卷目(如,VOL_001_Original Review, VOL_002_Round 2 Review)。出于相同的原因,认证人也必须提供他们的eCopy,刻录在单个的CD、DVD或闪存盘上。
虽然认证人是FDA的点对点联系人,并且是在eCopy有任何问题时被发送eCopy暂挂通知的一方,但各方都有责任满足eCopy的要求。
e. 如果这是一个捆绑式申请怎么办?
对于捆绑式申请,应有一个可应用于捆绑中所有申请的附函和eCopy的版本,捆绑中的每项申请不应有不同的附函。
除了签名的附函和适当的eCopy声明外,还应包括该申请中所有提交的清单或列表,作为捆绑申请的一部分。清单或列表应说明受到变更影响的每个器械的申请编号、商品名、以及型号(如适用)。
f. 如何向FDA提交eCopy?
eCopy与纸件申请同时提交。首先,将带有eCopy声明的签名附函作为eCopy的附件,然后将此eCopyn包装在纸质申请中,送至CDRH或CBER的DCC。无附函和纸质资料的eCopy将被列为eCopy暂挂。
如果提交了多份eCopy,则对所有eCopy提交一份附函即可。
对于第三方510(k)s,有关涉及双方各自的eCopy和附函的详情,请参见第7d节。
g. FDA如何处理eCopy?
eCopy是否加载成功由DCC确定,同时,FDA会收到该申请并录入我们的数据库。
如果eCopy加载成功,则附函和eCopy的内容将加载到相应中心的官方申请存储库中。
如果eCopy加载失败(即被拒绝),我们将以书面形式(如通过信函,电子邮件和/或传真)通知贵公司提交的eCopy暂挂。通知将说明eCopy加载失败的原因以及重新提交eCopy的渠道。请务必按照以下说明进行操作,以免在处理替换的eCopy时出现延迟。提交将被列为并维持在eCopy暂挂,直到向FDA提交有效的替换eCopy并经确认符合标准[1]。如果贵公司在180天内没有提供替换的eCopy,该提交将被视为撤回并在我们的数据库中关闭。
h. 如果贵公司的提交被列为eCopy暂挂,贵公司可向FDA提供什么?
如上所述,贵公司将收到一个通知,说明贵公司的eCopy加载失败的具体原因。为回复eCopy的斩挂通知,贵公司必须提供:(1)修订后的附函,对现在提供的替换eCopy 的附加声明和(2)一份替换的eCopy(CD,DVD或闪存盘)。
无论eCopy加载失败是否与附函相关,都需要修订附函,因为DCC需要能够对修订的附函加盖日期章以记录替换的eCopy的接收日期。需确保修订的附函包括了签名和适当的eCopy声明。
i. FDA如何处理替换的eCopy?
当FDA收到替换的eCopy时,其处理方式与原eCopy相同。更具体地说,确定替换的eCopy是否通过了加载过程。如果没有,那么该申请将再次被列为eCopy暂挂,并将向贵公司发出eCopy暂挂通知。
j. 对于所提交申请的审评何时开始?
直到收到有效的eCopy,并且用户费用已支付(如适用),否则审评不会开始。
此外,如果该申请类型适用于受理和/或入档审查,否则,该申请就开始实质性审查。[2]
k. 如果贵公司针对不需要eCopy的提交类型提交了eCopy,并且收到了eCopy暂挂信,贵公司可作何选择?
如果贵公司为关怀使用IDE、紧急使用IDE、EUA、MAF、513(g)或CLIA X文件提交了eCopy,且不符合附件1中的标准,贵公司的提交将被列为eCopy暂挂。但是,与要求有效eCopy的其他提交类型不同,可以选择使用一份额外的纸质副本来替代eCopy,作为对eCopy暂挂通知的回复。
8. 如果贵公司的器械受CBER监管怎么办?
a. 新的eCopy要求适用吗?
是的,除非贵公司的申请是根据本指南豁免的完全电子提交,如下所述。根据法定要求实施,无论FDA对提交申请进行审评的中心,第4节中列出的所有医疗器械申请类型必须附有eCopy,除非该要求可以免除或豁免。因此,根据FD&C法案进行审评以及通过向CBER的DCC提交纸件副本的申请均必须附有eCopy,除非有下述豁免情况。
虽然许多提交给CBER的申请仍然是纸质格式,并要求提交多个副本,CBER目前也能够接收和管理完全电子提交。
根据公共卫生服务(PHS)法案获得许可证的器械所提交的申请,包括生物制品的许可证申请和补充申请,研究性新药的申请,以及EUAs和这些器械的提交前申请,均可以提交完全电子申请,见以下第8b和8c节。FDA将对此类完全电子申请豁免eCopy的要求。
对于任何提交给CBER的完全电子提交申请,FDA还免除了其根据eCopy的要求提交的纸质副本。因此,在以下第8c节中确定的符合CBER指南的完全电子提交,不需要附有纸质副本。
b. 贵公司可以选择提交电子申请吗?
是的,当贵公司选择以电子方式提交申请时,对于企业和CBER的员工会有诸多益处。
对贵公司来说,主要优势是节约开支。与打印、装订、标签制作和运输多个纸件相关的成本会非常大,特别是对于含有大量支持文档的申请。同样,我们期望FDA认可财政节约,因为贵公司选择以电子方式提交时,FDA避免了与追溯、通路和存储大量纸张相关的成本。
使用电子提交程序的另一个优点是,提交和审评中涉及的所有各方都参考同一份文件 - 电子文件,而不会有纸质文件是否是eCopy的真实副本这种问题。电子提交还可以减少审评员由于无法阅读或解释纸件副本上的信息(有时会在复印时出现)而要求重新提交先前已提交的信息的要求。
c. 如何准备和向CBER提交电子申请?
对于选择电子提交的申请人,C BER有几个资源可供选择,请见文件“生物制品注册申请的电子格式提交”。(http://www.fda.gov/BiologicsBloodVaccines/DevelopmentApprovalProcess/ucm163685.htm)。因此,该引用的参考文献提供了具体细节,在本指南中不再重复。
对于PHS法案监管的器械,需要提交BLA,在准备电子提交时,详见题为“以电子格式向生物制品评审和研究中心(CBER)提交注册申请文件- 生物制品上市申请”的指南(http://www.fda.gov/downloads/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation / Guidances / General / UCM192413.pdf)。注意,本指南的某些部分,例如药理学和毒理学的部分,通常与已批准的器械无关。
为递交给CBER的其他器械申请而准备电子提交的指南(例如510(k),PMA),请参阅“工业指南:以电子格式提交注册申请- 一般考虑” (www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM072390.pdf) 和“CBER SOPP 8110:向CBER提交纸质版注册申请”(http://www.fda.gov/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/ ProceduresSOPPs / ucm079467.htm),其包括了向CBER提交电子副本的信息。
我们目前正在为呈送给CBER的其他电子提交制定另外的更新指南,并发布了经修订的、更新的指导文件草案,题为“工业指南(草案):以电子格式提交注册申请 - 一般考虑” (http://www.fda.gov/RegulatoryInformation/Guidances/ucm124737.htm),本文件将为申请人准备电子提交提供额外信息。
贵公司在准备电子格式提交时的相关问题,可发送至CBER:ESUBPREP@fda.hhs.gov.
贵公司也可以通过CBER.CDISC@fda.hhs.gov联系CBER,讨论以CDISC格式提交数据的可能性(http://www.fda.gov/BiologicsBloodVaccines/DevelopmentApprovalProcess/ucm209137.htm).
CBER将通过电子传输(即通过电子提交网关[3],[4])或通过CBER的文件控制中心接受物理介质的电子申请。
附件1 - eCopies标准
以下是写入FDA eCopy软件编码的标准。如果eCopy不符合以下A至D节中规定的所有必需标准,则eCopy将无法通过FDA的eCopy加载过程。另一方面,第E至G节给出了在准备eCopy时的建议。
A. 附函要求
除了所需的签名[5],为了满足eCopy程序的标准,附函还必须包括以下eCopy声明之一:
eCopy是纸质副本的完全复制品;或
eCopy是纸质副本的完全复制品,除了[说明所有差异]。
我们希望绝大多数申请的内容包括第一个声明。然而,如上文第3节所述,对于包含了某些临床数据集或其他类型的无法或不可能采用纸质形式提交的数据/信息,则适用于第二种声明。
如果提交中包含了第二种eCopy声明,则必须说明eCopy和纸质提交之间的差异。此外,必须在纸质提交中包括一个占位符,指向eCopy的特定信息(例如,“eCopy是纸件的完全复制品,除了数据行列表,其仅在eCopy中提供。在纸件的第4卷中提供了占位符,以回溯参考该信息的eCopy。”)
还请注意,对于在形式审查期间发现的缺陷,FDA希望贵公司不要将回复放在附函中,以便于处理,并最大限度地减少附函中涉及的纸件扫描,而是请贵公司将回复放在申请主文件中。
B. 成卷或非成卷形式的结构要求
eCopy的结构高度依赖于申请的整体大小,可以基于成卷或非成卷的申请形式进行整理,如下所述:
1. 成卷的eCopy
以卷为基础的eCopy通常推荐用于大的或复杂的申请,以使eCopy的结构与纸质版本相吻合,而便于该申请的审查。该eCopy结构包括根目录级的卷(即文件夹),每个卷依次包括一个或多个PDF文件。
需要对卷进行命名规定,以确保系统可以创建与提交的纸质版申请相匹配的文件夹的排列顺序。每个卷必须采用以下命名规定:
VOL_XXX(例如, VOL_001);或
VOL_XXX描述性名称(例如,VOL_001_患者研究数据)。
卷的编号必须是从VOL_001开始的不重复的连续编号,卷号上限为999。如果未遵循此项卷的命名规定,eCopy将加载失败。
对卷的描述性名称是可选的。但是,如果对卷使用了描述性名称,则卷名应描述其内容,以使审评员明白其意。描述性名称最多为250个字符,但不能包含特殊字符(例如,波形符(~),星号(*),斜杠(/),反斜杠(\),冒号(:),问号(?),单引号('),双引号(“),小于号(<),大于号(>)或竖线(|))。如果使用了这些特殊字符之一,eCopy将加载失败。
基于卷的eCopy结构的轻微变化,同时包括根目录级别的卷和PDF文件,这种结构通常发生在申请人在根目录级别增加附函的PDF时,而所有其他的PDF则被整理在许多卷中。
没有子文件夹:在此eCopy结构下,必须避免在卷目录下放入任何子文件夹,否则eCopy将加载失败。
2. 非成卷的eCopy
非成卷的eCopy通常仅推荐用于文件较小的申请,此eCopy结构是在根目录级别包括一个或多个PDF。
图1提供了以卷为基础的IDE示例。图2提供了包含多个PDF文件的非成卷的510(k)申请示例。
[1]请勿将“eCopy 暂挂”混同于FDA的决定,如拒绝受理或拒绝入档。eCopy 暂挂是发生在申请进行审评过程之前。一旦进入审评,受理和/或入档审查已完成(该申请类型如适用),亦可见本指南的第7.j. 部分。
[2]更多信息请参见指南“FDA和产业行动,上市前通告(510(k)) 申请:对FDA审评时钟和目标的影响”(http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/ucm089735.htm)和“工业和FDA工作人员指南- FDA 和产业行动,上市前批准申请(PMAs):对FDA审评时钟和目标的影响”(http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/ucm089733.htm).
[3] 25请参见“FDA 电子提交网关(联邦注册公告)- 8/9/2006”(http://www.gpo.gov/fdsys/pkg/FR-2006-08-08/html/E6-12808.htm).
[4]请参见“电子提交网关”(http://www.fda.gov/ForIndustry/ElectronicSubmissionsGateway/default.htm).
[5]贵公司还可以选择在eCopy中附上附函的电子版本;然而,这不应该代替所附的签名附函。
图1:成卷的研究性器械豁免(IDE)
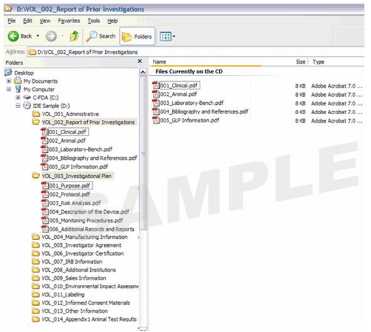
图2:非成卷的510(k)申请

C. Adobe Acrobat PDF文件要求
无论是选择成卷还是非成卷的eCopy结构,PDF文件都是用于eCopy的主要文件格式。(有关如何向eCopy添加非PDF文件,请参阅第D节。)PDF文件可确保提交时不会发生意外更改,并确保审评员在屏幕上看到的内容与纸质提交的内容相同。以下是对PDF文件的要求。若不遵守,贵公司的 eCopy将加载失败。
1. AdobeAcrobat PDF版本10.0或以下
只接受使用AdobeAcrobat 10.0或更低版本提交的eCopies。[1]如果贵公司的Adobe Acrobat是新版本(大于10.0),则必须将PDF另存为缩小的PDF,否则eCopy将无法加载。
2. 没有附件
eCopies提交的PDF文件若带有附件则不被接受,附件必须在提交之前删除。许多由第三方供应商创建的Acrobat PDF文件,如杂志文章,用户指南和产品标签,包含了在创建过程中嵌入的附件。例如,见下图3,其中包括名为joboptions的附件。要检测附件,请打开Acrobat PDF,然后单击导航面板左下角的回形针图标,如下图3所示。
图3:附件
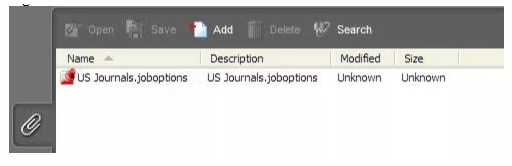
删除附件:
直接与供应商联系,要求他们创建AcrobatPDF,而不附加如上所示的任何附件; 或
打印选择Adobe PDF作为打印机名称的文件,该文件将打印成一个新的、没有附件的AcrobatPDF文件。
请注意,手动删除此附件(而不是按照上述步骤操作)则可能无法从文件中完全删除附件的属性,从而导致eCopy加载失败。
3. 无安全设置
不接受带有安全设置的PDF文件。PDF文件存储为原始文档,且不会更改原始格式。删除所有的安全设置,包括文件上使用的只读和密码保护。
FDA注意到,在我们网站上有某些表格设置了密码保护,我们目前正在用不含密码保护的版本替换用于上市前申请的一些主要表单。但是,对于仍有密码保护的FDA表单,贵公司当前可以选择的是:(1)填写表单,打印并扫描,或(2)将其保存在MISC FILES文件夹下的zip文件夹中,描述如下(第 D节)。
4. 具体的PDF文件命名规定
需要进行命名规定,以确保加载系统可以创建PDF文件的排列顺序,无论是否是成卷提交的部分,均应与提交的纸质版文件相匹配。所有PDF文件必须采用以下命名规定,无论是成卷或非成卷eCopy的部分:
XXX描述性名称(例如,001_附函,002_MDUFA 表,003_目录)。
PDF文件编号必须是不重复的、连续的3位数字编号,以001开头。如果未遵循此PDF文件命名规定,eCopy将加载失败。即使只有一个PDF文件,此命名规定也适用。
请记住,如果贵公司提交的申请是基于成卷形式,则需对每个卷中的PDF进行从头编号,如上图1所示。
描述性文件名最多可包含250个字符,但不能包含特殊字符(例如,波形(~),星号(*),斜杠(/),反斜杠(\),冒号(:),问号(?) ,单引号('),双引号(“),小于号(<),大于号(>)或竖线(|))。如果可能,文件名应描述其内容,以使审评员明白其意。如果使用了这些特殊字符之一,eCopy将加载失败。
5. PDF文件大小限制在50MB或以下
虽然对提交文件的总大小没有限制,但每个PDF文件必须限制为50MB或更小。
如果文件大于50MB,则必须将内容拆分为多个文件,并按命名顺序使用下一个文件编号,如下图4所示。如果必须拆分文件内容,建议对文件命名的方式应能清楚地反映其内容,一种方式是体现页码,如图4所示。
图4:PMA提交的示例,文件名中包含页码

B. 如何通过“统计数据”和“MISC文件”文件夹添加非PDF文件的要求
第B节中描述了成卷型和非成卷型的eCopy如何包括PDF文件,然而,eCopy还可以包括非PDF文件。这是通过在eCopy的根目录级别加入“统计数据”和/或“MISC FILES”文件夹来实现的,不论是成卷型或非成卷型的eCopy。
这两个文件夹是可选的; 但是,如果贵公司选择合并其中一个或全部两个,要求如下:
精确地拼写文件夹名称(即,统计数据,MISC文件),不区分大小写;
将文件夹放在eCopy的根目录级别;以及
将所有内容(请参阅下述第1和第2部分)列在文件夹下的一个或多个zip文件中。
如果不遵守这些要求,贵公司的eCopy将加载失败。
请注意,对于zip文件或添加到zip文件中的任何内容,没有命名规定的限制/限制条件。此外,zip文件没有大小限制;但是,建议贵公司将大小限制为50MB或更小,以避免潜在的问题,如上传文件时出现延迟。
对于适合作为“统计数据”或“MISC FILES”文件夹一部分的资料类型,描述如下。
1. “统计数据”文件夹
包括了元数据[2]和数据行列表的统计信息,可以以其原始格式包括在eCopy中,例如,但不限于:SAS;XPORT;XML;SGML;S- Plus;R files;ASCII;Molfiles 和Excel,对文件原格式没有限制。
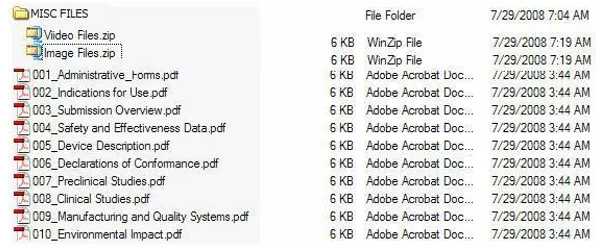
2. “MISCFILES”文件夹
有些申请可能需要提交杂项文件(例如,视频,X射线,机器可读的软件源代码),其不能以PDF格式提交(或不应提交)且不具有统计性质。这些杂项文件可以以其原始格式包含在MISCFILES文件夹下的eCopy中,例如但不限于:.gif;.tif;.jpg;.avi;.mpeg;.wmv;和.txt,对文件原格式没有限制。
此外,为了简化审评过程,FDA鼓励贵公司在MISC FILES文件夹下,列入某些文档或资料片段的Word版本,这些文档或资料片段也应以PDF格式在eCopy主体中提供。换句话说,将eCopy主体中的PDF版本作为成卷型或非成卷型的eCopy结构的一部分,并在MISC FILES文件夹中纳入Word版本,以助于审评。FDA通常会通过互动式审评要求下列文件,互动式审评使FDA可向申请人反馈和/或完成审评。将这些文件放入eCopy的MISC FILES文件夹中,有助于尽可能地减少对申请进行实审时的延误可能性。建议的文件包括,如适用:
任何申请的标签(最好是将每项(例如,给医生的标签,给患者的标签,操作员手册)作为单独文件];
510(k)s中与参比器械的对比表;
510(k)综述;
PMA的安全性和有效性数据(SSED)综述;以及
HDEs的安全性和可能的受益(SSPB)综述。
如上所述,对于添加到MISC FILES文件夹的zip文件中的任何文件,没有命名规定限制/限制条件。但是,当涉及MISC FILES文件夹中包含的Word文档时,建议使用与其PDF的等同性文件相同的命名,以使审评员易于将其关联。
如上述C.3节所述,FDA认识到,官网上有一些表格设有密码保护。一种选择是,将加入到eCopy中的具有密码保护的FDA表单,添加到MISC FILES文件夹下的zip文件中。
图5提供了具有统计数据文件夹的eCopy的示例。图6提供了具有MISCFILES文件夹的eCopy的示例。两者均直接反映了文件夹下的zip文件。
图5:具有统计数据文件夹的eCopy
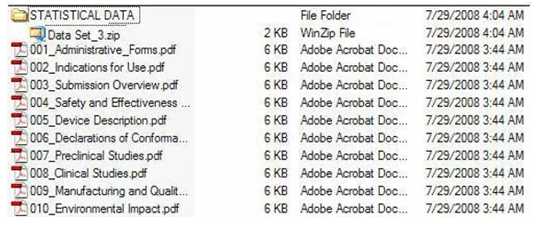
图6:具有MISC FILES文件夹的eCopy
C. PDF中的书签和超链接
应使用一个单独的PDF文件列入书签和超链接,以帮助审评员浏览申请的内容。如果使用了书签或超链接,请考虑以下事项:
1. 书签
可以为条目、子条目的标题,或者图形和表格的标题创建书签索引,做成一个单独的PDF文件。通常,为申请或项目的主目录加上一个有意义的书签是有益的,将有助于审评员对资料信息的定位和申请内容的导航。创建书签时,放大率设置应设为继承缩放,以使目标页面显示的倍率与审评员用于审阅文档其余部分的相同。
2. 超链接
鼓励采用超链接来改进对单个PDF文件的导航,超链接可以通过使用细线或蓝字的矩形来指定,或者可以在目录中使用不可见矩形的超链接,以避免其遮挡文本。在文件主体中的超链接用以支持不在同一页面上的注释、相关部分、参考文献、附录、表格或图形,并可提高导航效率。
当添加到中心的官方文档存储库时,在指定的PDF文件中,允许并维持在一个单独的PDF文档中其他部分的超链接。但是,不支持从一个PDF到另一个PDF文件(或跨其他文件类型)的超链接,该操作目前无法执行。
D. 从源文件创建PDF(首选)
从源文件(例如,Word文档)创建PDF时,请考虑以下事项:
1. Adobe插件
如果使用Adobe插件创建PDF文件和/或获得或显示数据,则可能有无法正确显示信息的风险,因为审评员可能无法访问某些插件以查看插件所显示的内容。
2. 字体
在源文件中应使用以下字体之一:TimesNew Roman; Verdana; Arial; Tahoma; 或Helvetica,应避免在同一文档中使用自定义字体和多种字体。
建议使用的字体颜色为黑色,超链接可使用蓝色字体。如果使用黑色以外的字体颜色,请避免使用在灰度打印机上无法正常打印的浅色。建议在提交前使用灰度打印机打印文档中的测试页来检查颜色的重现性。除了字体颜色,可采用表格以保持格式简单化。
E. 从扫描文档创建PDF文件
申请人应当在可行时直接从源文件创建所有PDF文件,而不是通过扫描创建。通过扫描纸质文件而生成的PDF文件远远不如从源文件直接生成的文档,例如Word文档,因此,如可能,应避免。扫描文档,特别是表格和图形,更难以读取,也不允许审评员复制和粘贴文本以进行编辑。
对于任何扫描的文档,应完成光学字符识别(OCR),以便文本可供搜索。通过以下方式检查内容是否已正确转换:(1)突出显示文本区域,(2)搜索单词或短语。如果在搜索中未搜到单词或短语,则OCR不能识别文本。FDA认为,OCR在某些情况下可能不适用于具有图表和图像的文档。
扫描的文档,特别是具有图像和照片的文档,往往文件较大,因此请注意,单个文件不要超过50MB,否则eCopy将加载失败。
请注意,FDA认为,可能有必要将扫描的文档添加到eCopy中。例如,如果贵公司没有对510(k)的真实性和准确性声明进行数字签名,那么该签名文档的扫描PDF副本应添加到eCopy中。
[1]一旦测试和确认Adobe Acrobat的新版本,我们将相应地更新该指南。
[2]元数据包括数据字典和术语,格式,注释的病例报告表,统计分析细节以及有助于理解和使用数据的任何其他信息。
eCopy Program for Medical Device Submissions Guidance for Industry and Food and Drug Administration Staff
Document issued on: December 31, 2012
The draft of this document was issued on October 17, 2012.
For questions regarding this document, contact CDRH’s Office of Device Evaluation at 301-796- 6055 or CBER’s Office of Communication, Outreach and Development at 1-800-835-4709 or 301-827-1800. U.S. Department of Health and Human Services Food and Drug Administration Center for Devices and Radiological Health Center for Biologics Evaluation and Research.
U.S. Department of Health and Human Services Food and Drug Administration Center for Devices and Radiological Health Center for Biologics Evaluation and Research
Preface
Public Comment
You may submit written comments and suggestions at any time for Agency consideration to the Division of Dockets Management, Food and Drug Administration, 5630 Fishers Lane, rm. 1061, (HFA-305), Rockville, MD, 20852. Submit electronic comments to http://www.regulations.gov. Identify all comments with the docket number listed in the notice of availability that publishes in the Federal Register. Comments may not be acted upon by the Agency until the document is next revised or updated.
Additional Copies
Additional copies are available from the Internet. You may also send an e-mail request to dsmica@fda.hhs.gov to receive an electronic copy of the guidance or send a fax request to 301- 827-8149 to receive a hard copy. Please use the document number (1797) to identify the guidance you are requesting. Additional copies of this guidance document are also available from the Center for Biologics Evaluation and Research (CBER), Office of Communication, Training and Manufacturers Assistance (HFM-40), 1401 Rockville Pike, Suite 200N, Rockville, MD 20852-1448, or by calling 1-800-835-4709 or 301-827-1800, by emailing ocod@fda.hhs.gov, or from the Internet at http://www.fda.gov/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/defaul t.htm.
eCopy Program for Medical Device Submissions 1 Guidance for Industry and Food and Drug Administration Staff
1. Introduction
The purpose of this guidance is to explain the new electronic copy (eCopy) Program for medical device submissions. Section 745A(b) of the Federal Food, Drug, and Cosmetic Act (FD&C Act), added by section 1136 of the Food and Drug Administration Safety and Innovation Act (FDASIA) (Pub. L. 112-144), requires the submission of eCopies with the issuance of this final guidance. This guidance describes how the Food and Drug Administration (FDA) is implementing the eCopy Program under section 745A(b) of the FD&C Act. The inclusion of an eCopy is expected to improve the efficiency of the review process by allowing for the immediate availability of an electronic version for review rather than relying solely on the paper version.
This guidance provides, among other things, the standards for a valid eCopy under section 745A(b)(2)(A) of the FD&C Act. In accordance with section 745A(b), submission types identified in this final guidance must include an eCopy in accordance with the standards provided by this guidance for the submission to be processed and accepted for review by FDA, unless they have been identified as being exempted or waived. Submissions submitted without an eCopy and eCopy submissions that do not meet the standards provided in this guidance will be placed on hold until a valid eCopy is submitted to FDA and verified to meet the standards, unless a waiver or exemption has been granted. While the submission is on hold, the review clock will not begin and the submission will not be reviewed.
In section 745A(b), Congress granted explicit statutory authorization to FDA to implement the statutory eCopy requirement by providing standards, criteria for waivers, and exemptions in guidance. Accordingly, to the extent that this document provides such requirements under section 745A(b) of the FD&C Act (i.e., standards, criteria for waivers, and exemptions), indicated by the use of the words must or required, this document is not subject to the usual restrictions in FDA’s good guidance practice (GGP) regulations, such as the requirement that guidances not establish legally enforceable responsibilities. See 21 CFR 10.115(d).
However, this document also provides guidance on FDA’s interpretation of the statutory eCopy requirement and the Agency’s current thinking on the best means for implementing other aspects of the eCopy program. Therefore, to the extent that this document includes provisions that are not “standards,” “criteria for waivers,” or “exemptions” under section 745A(b)(2), this document does not create or confer any rights for or on any person and does not operate to bind FDA or the public, but does represent the Agency’s current thinking on this topic. The use of the word should in such parts of this guidance means that something is suggested or recommended, but not required. You can use an alternative approach if the approach satisfies the requirements of the applicable statutes and regulations. If you want to discuss an alternative approach, contact the FDA staff responsible for implementing this guidance. If you cannot identify the appropriate FDA staff, call the appropriate number listed on the title page of this guidance.
To comply with the GGP regulations and make sure that regulated entities and the public understand that guidance documents are nonbinding, FDA guidances ordinarily contain standard language explaining that guidances should be viewed only as recommendations unless specific regulatory or statutory requirements are cited. FDA is not including this standard language in this guidance because it is not an accurate description of all of the effects of this guidance. This guidance contains both binding and nonbinding provisions. Insofar as this guidance provides “standards,” “criteria for waivers,” and “exemptions” pursuant to section 745A(b) of the FD&C Act, it will have binding effect.
The eCopy Program is not intended to impact (reduce or increase) the type or amount of data the applicant 2 1 includes in a submission to support clearance or approval. Please refer to other FDA device or program-specific guidance documents from CDRH (http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/defau lt.htm) and CBER http://www.fda.gov/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guida nces/General/ucm214106.htm) for the appropriate contents for submissions.
2. What is an eCopy?
An electronic copy (eCopy) is defined as an exact duplicate of the paper submission, created and submitted on a compact disc (CD), digital video disc (DVD), or a flash drive. An eCopy is accompanied by a paper copy of the signed2 cover letter and the complete paper submission.3 Note that inclusion of individual CDs for just certain sections of the paper submission (e.g., a CD of the data line listings added to a sleeve in some attachment) will no longer be accepted because such CDs will not meet the requirements for a valid eCopy as outlined in this guidance. Any CD provided must be an eCopy as defined above for the submission to be accepted.
3. Are differences between the contents of an eCopy and paper submission acceptable?
While an eCopy is defined as an exact duplicate of the paper copy, there are limited cases in which differences between the eCopy and the paper copy may be justified because a paper copy is not practical or appropriate for analysis purposes (e.g., raw data and statistical analysis programs, 3 4 data line listings to facilitate a bioresearch monitoring review) or is not feasible (e.g., videos, x-rays). The critical attribute of an eCopy is that it must include in electronic form all data required for that submission type. 5 In other words, the eCopy must include all of the required information for FDA review, whereas the paper copy can include a placeholder crossreferencing the location of certain information in the eCopy. The cover letter must contain the eCopy statement described in Attachment 1 and describe any differences between the paper version and the eCopy. If information is included in the eCopy that is not in the paper copy, then the paper copy must have a placeholder to redirect the reviewer (e.g., a piece of paper with a statement directing the reviewer to a particular section in the eCopy). FDA will consider the eCopy, with the cover letter, loaded into the appropriate Center’s official document repository to be the official record. Any undisclosed differences between the eCopy and the paper version may need to be rectified and could delay the review of the submission.
4. For what submission types is an eCopy required?
Section 745A(b) of the FD&C Act, as added by section 1136 of FDASIA, requires an eCopy for the following submission types:
· Premarket notification submissions (510(k)s), including third party 510(k)s;
· Evaluation of automatic class III designation petitions (de novos);
· Premarket approval applications (PMAs), including Transitional PMAs; 6
· Modular PMAs; · Product development protocols (PDPs);
· Investigational device exemptions (IDEs) [see partial exemption below];
· Humanitarian device exemptions (HDEs), including Humanitarian Use Device designation requests (HUDs);
· Emergency Use Authorizations (EUAs)7 [see exemption below];
· Certain investigational new drug applications (INDs);
· Certain biologics license applications (BLAs); and
· Pre-Submissions.
eCopies for all subsequent submissions to an original submission, including amendments (amendments include add-to-files and CLIA categorization add-to-files), supplements, and reports11 to the submission types identified above would also be required even if the original was submitted to FDA prior to implementation of the eCopy requirements.
The size of the submission is irrelevant. Whether it is a single-page submission (i.e., the cover letter is the only content) or a multi-volume submission, the eCopy requirements apply.
Responses to deficiency letters are required to be formally submitted to CDRH’s or CBER’s Document Control Center12 (DCC) to be logged in as an amendment or a supplement and, thus, are subject to the eCopy requirements.
Please note that eCopy requirements do not apply to information obtained during the Interactive Review process (via email, phone, and/or fax) once a submission is under review, if that information is not submitted to CDRH’s or CBER’s DCC. However, should an applicant choose to submit a response to an Interactive Review request to CDRH’s or CBER’s DCC (which should only occur if the size of the response makes communication by email or fax infeasible), it will be logged in as an amendment and be subject to the eCopy requirements.
Exemptions
Above, FDA identified the submission types cited in the legislation as being subject to the eCopy requirements. However, the legislation also allows for FDA to set forth criteria for exemptions from eCopy requirements. Accordingly, due to the potentially urgent nature of the following types of submissions, FDA considers these to be exempt from the requirement for an eCopy:
· two specific types of IDEs - compassionate use IDE submissions and emergency use IDE submissions; 5 13 and
· all EUAs.
Although these submission types do not require eCopies as per this exemption, FDA encourages you to submit eCopies of these submissions, when feasible, in order to facilitate the review process. If you choose to submit an eCopy, it must meet the standards outlined in Attachment 1.
Waivers
FDA believes that, given the widespread availability of software to enable the creation of an acceptable eCopy at little to no cost, all applicants should have the ability to provide an eCopy.
5. Are there other submission types not subject to the eCopy legislation for which eCopies may be submitted?
Although an eCopy is not required under Section 745A(b) of the FD&C Act, FDA also accepts and strongly encourages you to submit eCopies for:
· Master Access Files (MAFs);
· 513(g) Requests for Information (513(g)s); and
· Clinical Laboratory Improvement Act (CLIA) Categorization – Exempt Device submissions (CLIA “X” files).
eCopies for these three submission types are voluntary; however, if you choose to submit an eCopy, it must meet the standards outlined in Attachment 1.
6. How many copies of a submission are needed?
The eCopy Program does not change the overall number of copies to submit to FDA. Table 1 below provides the number of copies associated with each submission type for which an eCopy is either required or voluntary.
Table 1 – Number of Copies for Submission
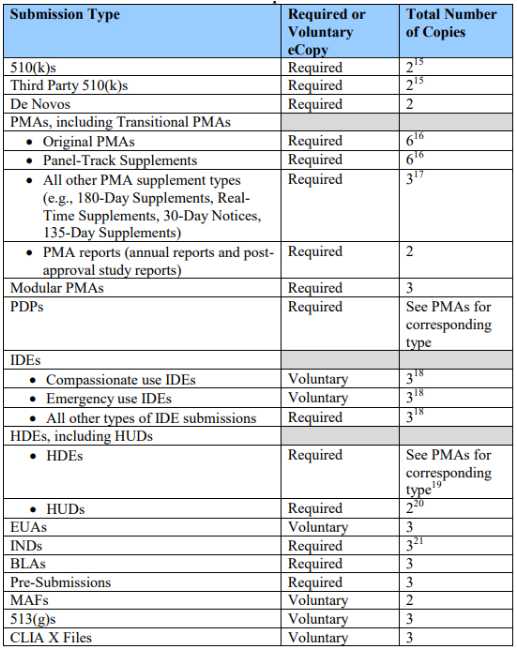
For those submission types for which an eCopy is required, an eCopy (with a signed cover letter and an adequate eCopy statement) will serve as one of the required number of copies for the applicable submission types. FDA will accept additional eCopies in lieu of additional paper copies as long as at least one paper copy is submitted along with the eCopy and the total number of required copies remains the same.
For submission types for which only two copies are required to be submitted, one must be an eCopy and the other must be a paper copy. For submission types requiring more than two copies, this policy would allow additional flexibility in the format of the application that is submitted. For example, for an original PMA, you would submit: (1) one eCopy and five paper copies; (2) five eCopies and one paper copy; or (3) any other combination that results in six total copies as long as there is at least one eCopy and one paper copy.
You may choose to submit only paper copies for compassionate use IDE submissions, emergency use IDE submissions, EUAs, MAFs, 513(g)s, and CLIA X files. However, if you choose to submit an eCopy for one of these, see above for details.
7. What are the processing steps for an eCopy?
Below are the processing steps for the submission and acceptance of an eCopy.
a. What are the standards for an eCopy?
With regard to the standards for an eCopy submitted to FDA, please refer to Attachment 1. An eCopy that does not meet the standards in Attachment 1 will fail to be accepted by our loading system.
b. How do you know if an eCopy meets the standards before you send it to FDA?
At this time, there is no FDA tool available to pre-validate an eCopy that has already been developed by an applicant. However, there is a new free eSubmitter-eCopies tool available on FDA’s website at http://www.fda.gov/ForIndustry/FDAeSubmitter/ucm317334.htm, which we strongly encourage applicants to use. Use of this tool is optional; however, one of the benefits of the tool is that it creates an eCopy in real-time that is consistent with the standards described in Attachment 1. Use of the eSubmitter-eCopies tool is intended to prevent delays in review of your submission due to the need to resolve technical issues.
Please note that this tool provides the formatted eCopy content for you to download onto your local drive and then burn onto a CD, DVD, or flash drive. There is no transmission of an eCopy through the FDA Gateway. Should you have any technical questions when generating your eCopy, please contact cdrhesub@cdrh.fda.gov prior to submission of the 8 eCopy to FDA.
c. What if there is another processing party involved?
In the case that another party (e.g., law firm, consultant) submits a submission on behalf of an applicant, the eCopy must still meet the standards for an eCopy in order to be successfully processed, whether accomplished by you (the applicant) or the submitting party. While the applicant may or may not include their own cover letter as part of the eCopy, our standards require that the submitting party include a signed cover letter with an eCopy statement, as described in Attachment 1.
d. What if this is a Third Party 510(k)?
There are two distinct parties involved in the generation of a Third Party 510(k): (1) the Accredited Person and (2) the applicant. Each party is subject to the eCopy requirements. Accordingly, each party (i.e., the Accredited Person and the applicant) must provide their own:
· eCopy (on a single CD, DVD, or flash drive) that meets the standards in Attachment 1; and
· signed cover letter with an eCopy statement as described in Attachment 1.
Therefore, there will be two separate eCopies provided for a given Third Party 510(k). Given this, it is essential that each eCopy be clearly marked as belonging to the Accredited Person or the applicant.
FDA recognizes that an applicant may interact with the Accredited Person multiple times prior to the Third Party 510(k) being submitted to FDA. Regardless of the number of interactions prior to the Third Party 510(k) being submitted to FDA, the applicant’s eCopy must be on a single CD, DVD, or flash drive in order to be loaded by our software. This could be accomplished by, for example, organizing each round of interaction as different volumes in the eCopy (e.g., VOL_001_Original Review, VOL_002_Round 2 Review). The Accredited Person must also provide their eCopy on a single CD, DVD, or flash drive for the same reason.
Although the Accredited Person is FDA’s point-of-contact and is the party that will be sent the eCopy hold notification if there are any issues with either eCopy, each party is responsible for meeting the eCopy requirements.
e. What if this is a bundled submission?
For bundled submissions, there should be one version of the cover letter and the eCopy that applies to all submissions in the bundle. There should not be different cover letters associated with each submission in the bundle.
In addition to the cover letter being signed and having an adequate eCopy statement, it should include a list or table of all submissions that are part of the bundle. The list or table should specify the submission number, trade name, and, as applicable, the model number, of each device impacted by the change.
f. How do you submit an eCopy to FDA?
An eCopy is submitted simultaneously with the paper submission(s). First, attach the signed cover letter with the eCopy statement to your eCopy. Then attach this eCopy package to the paper submission(s) and send them to CDRH’s or CBER’s DCC. An eCopy that is sent to the DCC without a cover letter and accompanying paper submission(s) will be placed on eCopy hold. If more than one eCopy is to be submitted, then a single cover letter for all eCopies is sufficient. For Third Party 510(k)s, refer to Section 7d for the specifics of the eCopy and cover letter associated with each of the two parties involved.
g. How does FDA process an eCopy?
The determination as to whether or not an eCopy passes the loading process will be made by DCC at the same time the submission is received by FDA and logged into our database. If an eCopy passes the loading process, the cover letter and eCopy contents will be loaded into the appropriate Center’s official submission repository. If an eCopy fails the loading process (i.e., is rejected), we will notify you in writing (e.g., by letter, email, and/or fax) that your submission is on eCopy hold. The notification will describe the reasons for the eCopy failure and the logistics for submitting a replacement eCopy. It is important that you follow these directions to avoid delays in processing the replacement eCopy. The submission will be placed and remain on eCopy hold until a valid replacement eCopy is submitted to FDA and verified to meet the standards. 9 23 If you do not provide a replacement eCopy within 180 days, the submission will be considered withdrawn and closed in our database.
h. What do you provide to FDA if your submission was placed on eCopy hold?
As stated above, you will receive a notification that states the specific reason(s) why your eCopy failed the loading process. In response to the eCopy hold notification, you must provide: (1) a revised cover letter that now additionally states you are providing a replacement eCopy and (2) a replacement eCopy (CD, DVD, or flash drive).
The revised cover letter is required regardless of whether or not the eCopy failure was related to the cover letter, because the DCC needs to be able to date stamp the revised cover letter to record the receipt date of the replacement eCopy. Be sure that your revised cover letter includes a signature and an adequate eCopy statement.
i. How does FDA process a replacement eCopy?
When FDA receives a replacement eCopy, it is processed in the same manner as the initial eCopy. More specifically, a determination is made as to whether or not the replacement eCopy passes the loading process. If it does not, then the submission will be placed on eCopy hold again, and an eCopy hold notification will be issued to you.
j. When does review of a submission begin?
Review of a submission will not begin until a valid eCopy has been received and, if applicable, the user fee has been paid. Furthermore, if applicable for that submission type, acceptance and/or filing reviews will be conducted. Otherwise, the substantive review of the submission will begin.
k. If you submited an eCopy for a submission type that did not require an eCopy and you received an eCopy hold letter, what are your options?
If you submitted an eCopy for a compassionate use IDE, emergency use IDE, EUA, MAF, 513(g), or CLIA X file, and it did not meet the standards in Attachment 1, your submission will be placed on eCopy hold. However, unlike the other submission types that require a valid eCopy, you have the option of responding to that eCopy hold notification with an additional paper copy in lieu of a replacement eCopy.
8. What if your device is regulated by CBER?
a. Will the new eCopy requirements apply?
Yes, unless your submission is an entirely electronic submission exempted under this guidance, as described below. Upon implementation of the statutory requirement, all medical device submission types listed in Section 4 must be accompanied by an eCopy regardless of the Center in FDA in which the submission will be reviewed unless the requirement is waived or exempted. Accordingly, submissions for devices subject to review under the FD&C Act and submitted by filing paper copies with CBER’s DCC must be accompanied by an eCopy, except where exempted as described below.
While many submissions made to CBER are still in paper format and require submission of multiple copies, CBER is also currently able to receive and manage submissions that are entirely electronic.
Submissions for devices that are subject to licensure under the Public Health Service (PHS) Act, including biologics license applications and supplements, investigational new drug applications, and EUAs and pre-submissions for these devices, may be submitted as entirely electronic submissions as detailed in Sections 8b and 8c below. FDA will exempt such entirely electronic submissions from the eCopy requirement.
FDA additionally waives the eCopy requirement to submit paper copies of any entirely electronic submission made to CBER. Accordingly, entirely electronic submissions that comply with CBER guidance identified in Section 8c below do not need to be accompanied by paper copies.
b. Can you submit an electronic submission instead?
Yes, and there are several advantages for both industry and for CBER staff when you choose to make submissions electronically.
The main advantage to you is in the financial savings that will likely result. The costs associated with printing, binding, labeling, and shipping multiple paper copies can be significant, especially for submissions that contain a great deal of supporting documentation. Likewise, we anticipate that FDA will recognize financial savings in that FDA avoids the costs associated with tracking, routing, and storing large amounts of paper when you choose to submit electronically.
Another advantage with the use of the electronic submission process is that all parties involved in the submission and review are referencing the same document – the electronic one. There is no question about whether the paper copy is an exact copy of the eCopy. Electronic submissions may also reduce the need for reviewers to request re-submission of previously submitted information due to an inability to read or interpret the information on the paper copy, as sometimes occurs when documents are photocopied.
c. How do you prepare and submit an electronic submission to CBER?
CBER has several resources available to applicants who choose to submit electronic submissions as outlined in the document “Regulatory Submissions in Electronic Format 11 for Biologic Products.” (http://www.fda.gov/BiologicsBloodVaccines/DevelopmentApprovalProcess/ucm163685 .htm). Thus, specific details are available in the cited references and will not be repeated in this guidance.
For devices that are regulated under the PHS Act and require the submission of a BLA, consult the guidance document entitled “Providing Regulatory Submissions to the Center 12 for Biologics Evaluation and Research (CBER) in Electronic Format - Biologics Marketing Applications” (http://www.fda.gov/downloads/BiologicsBloodVaccines/GuidanceComplianceRegulator yInformation/Guidances/General/UCM192413.pdf) for details on preparing your electronic submission. Note that certain sections of this guidance, for example, those on pharmacology and toxicology, are generally not pertinent to licensed devices.
For guidance on preparing electronic submissions for other device submissions (e.g., 510(k)s, PMAs) sent to CBER, please see “Guidance for Industry: Providing Regulatory Submissions in Electronic Format - General Considerations” (www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ UCM072390.pdf) and “CBER SOPP 8110: Submission of Paper Regulatory Applications to CBER” (http://www.fda.gov/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformatio n/ProceduresSOPPs/ucm079467.htm), which includes information about providing electronic copies to CBER.
We are currently developing additional, updated guidance for other electronic submissions sent to CBER and have issued a revised, updated draft guidance document for comment entitled, “Draft Guidance for Industry: Providing Regulatory Submissions in Electronic Format-General Considerations” (http://www.fda.gov/RegulatoryInformation/Guidances/ucm124737.htm). This document will provide an additional resource for applicants preparing electronic submissions.
You can submit questions pertaining to the preparation of submissions in electronic format for submission to CBER at ESUBPREP@fda.hhs.gov.
You may also contact CBER at CBER.CDISC@fda.hhs.gov to discuss the potential for submission of data in CDISC format (http://www.fda.gov/BiologicsBloodVaccines/DevelopmentApprovalProcess/ucm209137 .htm).
CBER will accept electronic submissions via electronic transmission (i.e., through the Electronic Submissions Gateway25,26) or on physical media through CBER’s Document Control Center.
来源:CMDE
整理:致众TACRO
相关文章
医疗器械的电子副本提交程序,行业和食品药品监督管理局工作人员指南
FlashFXP绿色版网盘下载,附激活教程 3396
FlashFxp百度网盘下载链接:https://pan.baidu.com/s/1MBQ5gkZY1TCFY8A7fnZCfQ。FlashFxp是功能强大的FTP工具
Adobe Fireworks CS6 Ansifa绿色精简版网盘下载 3321
firework可以制作精美或是可以闪瞎眼的gif,这在广告领域是需要常用的,还有firework制作下logo,一些原创的图片还是很便捷的,而且fireworks用法简单,配合dw在做网站这一块往往会发挥出很强大的效果。百度网盘下载链接:https://pan.baidu.com/s/1fzIZszfy8VX6VzQBM_bdZQ
navicat for mysql中文绿色版网盘下载 2930
Navicat for Mysql是用于Mysql数据库管理的一款图形化管理软件,非常的便捷和好用,可以方便的增删改查数据库、数据表、字段、支持mysql命令,视图等等。百度网盘下载链接:https://pan.baidu.com/s/1T_tlgxzdQLtDr9TzptoWQw 提取码:y2yq
火车头采集器(旗舰版)绿色版网盘下载 3167
火车头采集器是站长常用的工具,相比于八爪鱼,简洁好用,易于配置。火车头能够轻松的抓取网页内容,并通过自带的工具对内容进行处理。站长圈想要做网站,火车头采集器是必不可少的。百度网盘链接:https://pan.baidu.com/s/1u8wUqS901HgOmucMBBOvEA
Photoshop(CS-2015-2023)绿色中文版软件下载 3135
安装文件清单(共46G)包含Window和Mac OS各个版本的安装包,从cs到cc,从绿色版到破解版,从安装文件激活工具,应有尽有,一次性打包。 Photoshop CC绿色精简版 Photoshop CS6 Mac版 Photoshop CC 2015 32位 Photoshop CC 2015 64位 Photoshop CC 2015 MAC版 Photoshop CC 2017 64位 Adobe Photoshop CC 2018 Adobe_Photoshop_CC_2018 Photoshop CC 2018 Win32 Photoshop CC 2018 Win64